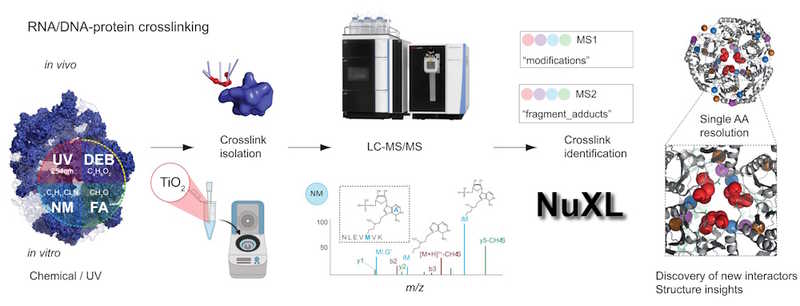
Стандартный метод для анализа взаимодействий «РНК–белок» объединяет ультрафиолетовые (УФ) кросс-сшивки и масс-спектрометрический анализ. Однако УФ-сшивки неодинаково работают с разными азотистыми основаниями — они эффективны для пиримидинов, но хуже срабатывают для пуринов или оснований двуцепочечной ДНК. Чтобы усовершенствовать методику анализа ДНК-белковых взаимодействий, авторы статьи в Nucleic Acids Research дополнили индуцированные ультрафиолетом кросс-сшивки химическими. Для анализа таких данных они также разработали NuXL — высокоспециализированную поисковую машину на базе открытого программного обеспечения OpenMS, который специфически распознает пептиды, сшитые с (олиго)нуклеотидами. Предложенный подход позволяет эффективно выявлять перекрестные сшивки РНК/ДНК с белками с разрешением на уровне отдельных аминокислот.
Разработка метода осуществлялась в несколько этапов. Авторы начали с хорошо охарактеризованных комплексов «белок-РНК» и «белок-ДНК», на которых отрабатывали и калибровали методику сшивания. В качестве химических сшивающих агентов ученые применяли диэпоксибутан, мехлоретамин (алкилирующий агент, также известный как хлорметин) и формальдегид. Одной из классических моделей стали рибосомы E. coli; для изучения белков с неструктурированными регионами, участвующими в связывании нуклеиновых кислот, авторы использовали комплекс фактора регуляции элонгации NELF с РНК. Модельной системой для изучения взаимодействий гистонов с ДНК служили нуклеосомы дрожжей S. cerevisiae.
Для сшивания на комплексы действовали УФ-излучением и разными концентрациями химических сшивателей. Несшитые РНК/ДНК участки разрушали до олигонуклеотидов нуклеазами, а методом хроматографии удаляли свободные нуклеотиды. После этого на шариках из диоксида титана (TiO₂) проводили обогащение — на них улавливали пептиды, сшитые с олигонуклеотидами (информацию о сайте связывания несут именно они). Полученные образцы анализировали методом масс-спектрометрии.
Экспериментальная часть включала также работу с целыми бактериальными клетками — культуру E. coli подвергали действию УФ-излучения и химических сшивателей. После этого для выделения нуклеиновых кислот клетки лизировали и разделяли лизат на фракции (S30/S100). Дальнейшая пробоподготовка была аналогична таковой для отдельных белковых комплексов — она включала расщепление свободных нуклеиновых кислот и обогащение, а также содержала дополнительные этапы очистки. Перед масс-спектрометрией, для уменьшения сложности смеси, пептиды разделяли на фракции с помощью высокоэффективной жидкостной хроматографии.
Отдельным этапом стала разработка NuXL. Эта поисковая машина предназначена для автоматизированного поиска в данных масс-спектрометрии пептидов, сшитых с нуклеотидами. Заранее заданные в программе пресеты содержат информацию о сдвигах масс, вызванные формированием сшивки с нуклеотидами под действием УФ или химических агентов. Сравнивая экспериментальные спектры с теоретическими, NuXL определяет пептидную последовательность, олигонуклеотидную последовательность, а также конкретную аминокислоту и нуклеотид, которые образовали сшивку.
 Credit:
Credit: Разработчики сравнили производительность NuXL с устоявшимся пайплайном, который основан на MSFragger. Метод верифицировали на известных структурах — рибосомах E. coli, пробоподготовка для которых описана выше. NuXL продемонстрировал более высокую производительность в сопоставлении спектров кросс-сшивок (CSM), а также показал преимущество в скорости анализа.
Затем авторы провели биоинформатический и структурный анализ, в результате которого идентифицировали тысячи сайтов сшивания. Картируя сайты сшивания на известные 3D-структуры рибосом, ученые подтвердили, что они обнаруживаются в местах контакта с нуклеиновыми кислотами, что подтверждает работоспособность метода. Также исследователи провели структурное моделирование — данные химического сшивания для комплекса ДНК-метилтрансферазы 2 (Dnmt2) с тРНК вместе с предсказаниями AlphaFold 3 использовали для создания точной структурной модели.
Всего с помощью NuXL авторы идентифицировали в протеоме E. coli более 1500 белков, взаимодействующих с нуклеиновыми кислотами, и более 4900 уникальных пептидов, формирующих кросс-сшивки.
При помощи статистического анализа ученые показали, что процесс сшивки не случаен: УФ специфично присоединяет нуклеотиды к тирозину и цистеину, а химические агенты — к метионину и глутамату.
Применив анализ генной онтологии, исследователи определили процессы и функции, в которых участвуют выявленные белковые молекулы. Они классифицировались не только как РНК-связывающие белки, участвующие в трансляции и сплайсинге, — среди них обнаружились также практически все ферменты гликолиза и цикла Кребса. Это подтверждает многофункциональность метаболических ферментов — они участвуют в регуляции на уровне РНК. Наконец, анализ ДНК-связывающих белков выявил, что транскрипционные факторы и белки нуклеоида также образуют сшивки с РНК. Предполагается, что связывание с РНК может быть свойством ДНК-связывающих белков (транскрипционных факторов), что открывает новое направление для исследований регуляции генов.
Авторы отмечают, что химическое сшивание не заменяет, а дополняет УФ-сшивки, позволяя получить более полную информацию. Идею этой разработки объясняет для PCR.NEWS Сергей Мошковский, научный сотрудник Института мультидисциплинарных естественнонаучных исследований Макса Планка в Геттингене (Германия).